Spatial transcriptOmics Analysis Resource
Citation
SOAR elucidates disease mechanisms and empowers drug discovery through spatial transcriptomics
Yiming Li, Saya Dennis, Meghan R. Hutch, Yanyi Ding, Yadi Zhou, Yawei Li, Maalavika Pillai, Sanaz Ghotbaldini, Mario Alberto Garcia, Mia S. Broad, Chengsheng Mao, Feixiong Cheng, Zexian Zeng, Yuan Luo
bioRxiv 2022.04.17.488596; doi: https://doi.org/10.1101/2022.04.17.488596
Contact
Yuan Luo, PhD: yuan.luo@northwestern.edu
Zexian Zeng, PhD: zexianzeng@pku.edu.cn
Data Browser
- Click on a Sample number to visualize spatial gene expression and view its spatial variability analysis results
- The datasets are categorized into different research topics (the Topic column), and sample-wise conditions are recorded in the Condition column
Loading...
Spatial variability analysis results
Loading...
Gene & Cell Analysis
Spatial variability
- Please select a gene.
Neighborhood-based cell-cell interaction
- Please first select a gene and next an available cell type.
- This function visualizes a gene’s differential expression between the query cell type capture locations adjacent/nonadjacent to interacting cell types.
Distance-based cell-cell interaction
- Please select (1) a distance threshold, (2) a signaling pathway, and (3) a query cell type
- This function models the interactions between different cell types through a given pathway
- Ligand-receptors paris included in pathways are derived from CellChatDB
Drug Discovery
- The pathological sample browser below displays the samples with significantly enriched CMAP compound instances
- CMAP is a large perturbational dataset published by Broad Institute, with the goal of providing a holistic understanding of the effect of compounds on cellular signatures via a high-throughput gene expression profiling technology. SOAR utilizes the perturbation data from CMAP to identify potential therapeutics for pathological samples in our database.
- The pathological sample browser have been pre-filtered to include perturbations with significant p-values and ranked within the top and bottom 50 perturbation instances in terms of enrichment score. The drug enrichment table include the top and bottom 500 significant perturbation instances.
- A positive enrichment score means the compound will have a suppressing effect on the cell’s spatially variable and differentially expressed genes and vice versa.
- Click on a Sample number to investigate each sample’s differential gene expression, protein-protein interaction network, drug enrichment, and drug perturbation network
Pathological Sample Browser
Loading...
*To download the full top/down 5,000 perturbation results table for exploration, please click https://vfsmrshiny04.fsm.northwestern.edu/skycurlapp/
Differential Gene Expression
Loading...
Protein-Protein Interaction
Loading...
Drug Enrichment
Inversely enriched (repress spatially variable DEGs)
Loading...
Positively enriched (enhance spatially variable DEGs)
Loading...
Drug Perturbation Network
Loading...
The raw data source can be accessed via the “source” link in the Data Browser module. All cleaned data used for analyses are available for download through the Data Browser and Drug Discovery modules.
Introduction
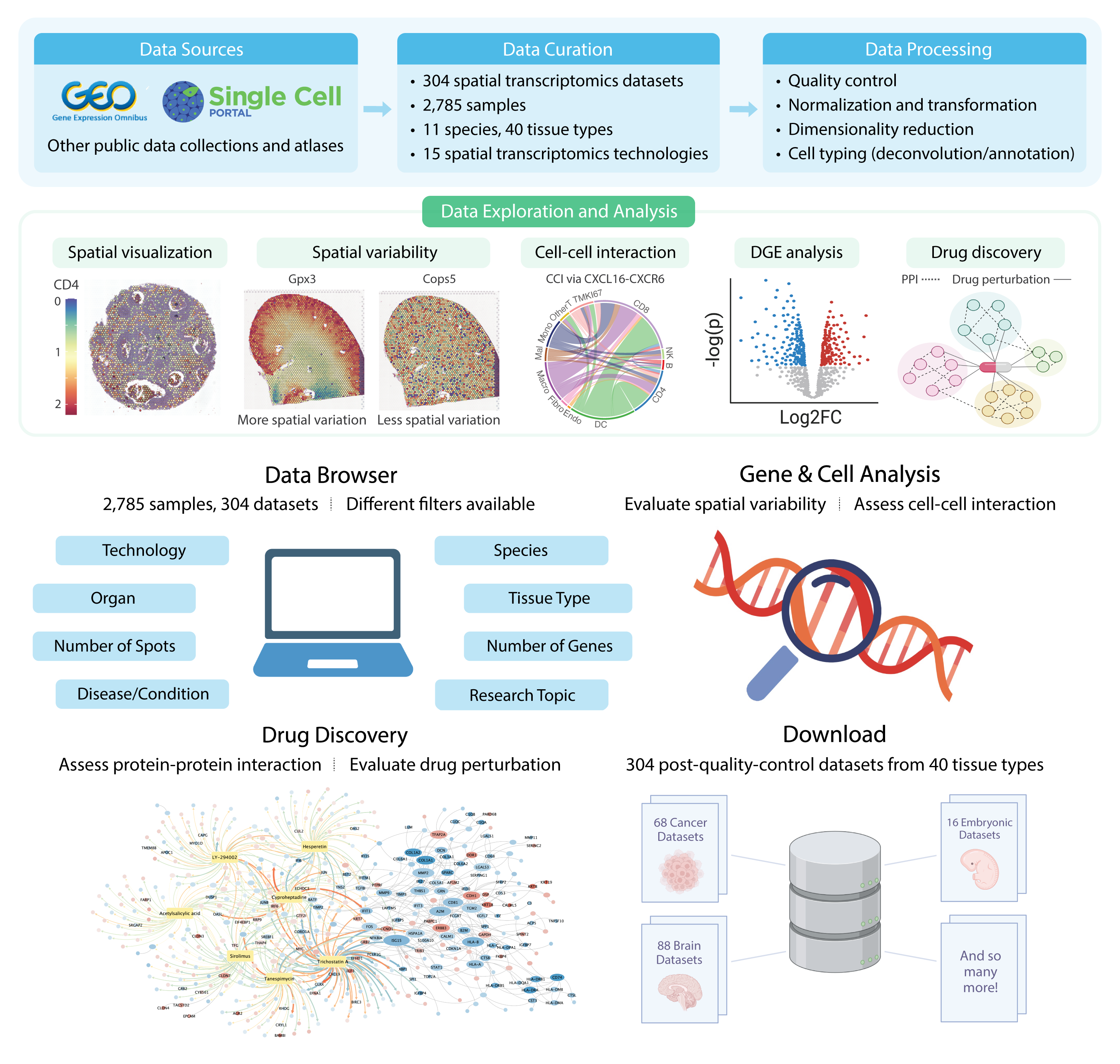
SOAR: Spatial transcriptOmics Analysis Resource is a database with an extensive collection of spatial transcriptomics (ST) data and analysis capability. ST can measure the transcriptome while preserving the spatial organization of capture locations. This allows researchers to study tissue functions and disease pathology in their morphological context. Large volumes of spatial transcriptomics data have been generated, but the lack of a large-scale database with systematically collected and processed data makes data reuse challenging. To facilitate data exploration and meta-analysis in ST research, we present SOAR, which provides unified data access, exploration, and analysis across different tissue types.
Tutorial
Please view a step-by-step tutorial <a href=https://drive.google.com/file/d/1F5_1oaSB5a7VonFk-8ZCDKb1Y4xCaGdZ/view?usp=sharing>Here.
Usage
SOAR is a comprehensive spatial transcriptomics database with biological insights exploration and drug discovery capabilities (Supplementary Fig. 1). This manual will demonstrate the interactive analysis functions offered by SOAR.
Data Browser
The Data Browser module contains the meta-data for all 3,461 ST samples on SOAR. Users can pinpoint their sample of interest using different filters, including the type of ST technology used (e.g. 10x Visium, Slide-seq, etc.), species, organ, tissue type, tissue condition, or research topic (developmental or pathological). We also document the average number of capture locations and genes in each sample after quality control.
After identifying a sample of interest, users can click on its sample number, and they will be directed to the spatial expression visualization panel. Users can then visualize the spatial expression of different genes in that sample, as well as view its annotated/deconvoluted cell types, spatial clustering, and spatial variability analysis results.
Gene & Cell Analysis
In the Gene & Cell Analysis module, the user can select a tissue type and species and next query their genes of interest. SOAR allows users to perform three types of analyses: spatial variability, neighborhood-based cell-cell interaction, and distance-based cell-cell interaction.
Spatial Variability
To facilitate the characterization of the functional architecture of complex tissues, we identified genes with spatial patterns of significant expression variation using SpatialDE. Spatial variability analyses were conducted across the whole tissue and in different cell types, respectively.
Neighborhood and Distance-based Cell-Cell Interaction
Cells of different cell types may interact through cell-cell contact or long-distance signalling. ST enables us to study cell-cell interactions by investigating whether a gene’s expression appears to be promoted or inhibited when in proximity to another cell type. To identify possible cell-cell interactions, we investigated whether the gene expression levels in a query cell type (\(CT_Q\)) are influenced by its spatial proximity (in terms of neighborhood or distance) to another cell type (\(CT_I\)).
In order to evaluate neighbouring interactions, we performed Wilcoxon rank-sum tests to test if genes are differentially expressed in \(CT_Q\) adjacent and nonadjacent to \(CT_I\) using the FindAllMarkers function in Seurat. If a gene \(G\) is more highly expressed in \(CT_Q\) adjacent to \(CT_I\), \(CT_I\) capture locations may promote the expression of \(G\) in \(CT_Q\).
To assess cell-cell interactions over distance, we evaluated the levels of cell-cell interactions through different signaling pathways in the CellChatDB database using COMMOT, an optimal-transport-based approach. For multiple-cell resolution data, the pseudo-cell-level deconvoluted expression data was used. In order to characterize the cell-cell interactions over different distances, we performed COMMOT analysis using short (500 μm), medium (1,000 μm), and long (1,500 μm) distance thresholds.
Drug Discovery
In the Drug Discovery module, the user may explore the drug screening results in samples of interest using the pathological sample browser. Four analysis modules are available, including differential gene expression, protein-protein interaction, drug enrichment, and drug perturbation network, providing dynamically generated visualizations for download.
Differential Gene Expression
We performed Wilcoxon rank-sum tests to test if genes are differentially expressed in a certain cell type compared with other cell types. For multiple-cell resolution data, the pseudo-cell-level deconvoluted expression data was used. The testing was limited to genes with at least 0.1 log fold change between two groups of cells and detected in a minimum fraction of 0.1 cells in either group.
Protein-Protein Interaction
For each cell type, we extracted the associated protein-protein interaction (PPI) modules using a maximum of 100 most significant differentially expressed genes and our human protein-protein interactome.
Drug Enrichment
To identify drug candidates with the potential for disrupting dysregulated processes in pathological samples, we conducted in silico drug repurposing by performing gene set enrichment analyses on the differentially expressed gene sets associated with each cell type. We retrieved the Connectivity Map L1000 dataset (accessed in October 2023) and analyzed perturbation profiles of 27,669 compounds associated with gene expression changes in 12,328 genes. These chemical perturbagens were treated in various cell lines and doses, resulting in a total of 145,491 perturbation profiles. Enrichment analyses were performed on the DEG sets with at least ten significantly (adjusted p-value < 0.05) up-regulated (log fold change > 0.5) or down-regulated (log fold change < -0.5) genes, and fewer than 1,000 total significant genes. We computed an enrichment score for each eligible differentially expressed gene set and Connectivity Map L1000 signature using the method described in Sirota et al. (2011).
Drug Perturbation Network
For each set of drug enrichment analysis results, the top 100 inversely related (enrichment score > 0, adjusted p-value < 0.05) and top 100 positively related (enrichment score < 0, adjusted p-value < 0.05) drugs were used to generate the drug perturbation network. In the network, each node is a drug or gene, and each edge reflects an identified perturbation of the drug on the gene. The gene nodes are colored by their log fold changes in differential gene expression analysis, and the edges are colored by the standardized perturbation scores.
Download
In the Download module, users can download all the processed gene expression data (gene by capture location), coordinates data (capture location by Cartesian coordinates), metadata, and sample-wise analysis results. We also provide the raw data for download in the Data Browser module.
Powered by RShiny. Visualizations created using ggplot2 and Seurat. Illustrations created using Adobe Illustrator and BioRender.com.
Citation
SOAR elucidates disease mechanisms and empowers drug discovery through spatial transcriptomics
Yiming Li, Yanyi Ding, Saya Dennis, Meghan R. Hutch, Jiaqi Zhou, Yadi Zhou, Yawei Li, Maalavika Pillai, Sanaz Ghotbaldini, Mario Alberto Garcia, Mia S. Broad, Chengsheng Mao, Parambir S. Dulai, Feixiong Cheng, Zexian Zeng, Yuan Luo
Science Advances (2025); doi: https://doi.org/10.1126/sciadv.adt7450
Our Mission
Recent technological advances in spatial transcriptomics (ST) have made it possible to measure the transcriptome while retaining the coordinates of capture locations in tissues. Spatially resolved transcriptomics allows researchers to study the association between gene expression and the spatial organization of capture locations.
The increasing utilization of such technologies by academic researchers propelled us to create a comprehensive ST database, Spatial TranscriptOmics Analysis Resource (SOAR).
Users may use SOAR to evaluate the spatial variability of genes, assess cell-cell interactions, perform drug discovery, as well as visualize spatial gene expressions in an interactive manner, and effortlessly “SOAR” into the richness of information that ST data holds to drive new biomedical discoveries.
Our Labs
The Luo Lab, located at Northwestern University’s Feinberg School of Medicine, is broadly interested in the research of machine learning, natural language processing, time series analysis, integrative genomics and computational phenotyping, with a focus on medical and clinical applications. Some of our recent works are on subgraph mining and factorization models applied to clinical narrative text, ICU physiologic time series and computational genomics. The common theme of these works aims at building clinical models that improve both prediction accuracy and interpretability by exploring relational information in each data modality.
The Zeng Lab, located at Peking University, focuses on developing algorithms and tools to help understand cancer immunology and immunotherapy, cell signaling, and gene expression regulation. Zeng lab is developing a number of algorithms and tools to analyze high throughput genomics data, including RNA-seq, WES, CRISPR Screen, single cell, spatial transcriptomics, ATAC-seq data, etc. Through data integration and algorithm development, Zeng lab tries to model the specificity and function of tumor development, progression, drug response, and resistance. Zeng lab also designs experiments on the bench hoping to understand the underlying mechanisms when data lead them to unexpected but interesting results.
Contact
Yuan Luo
750 N. Lake Shore Drive, 11/F
Chicago, IL 60611
Phone: 312-503-5742
Email: yuan.luo@northwestern.edu
—
Zexian Zeng
Lui Che Woo Building 235, Peking University, Haidian
Beijing, China 100084
Phone: +86-(01)62750092
Email: zexianzeng@pku.edu.cn